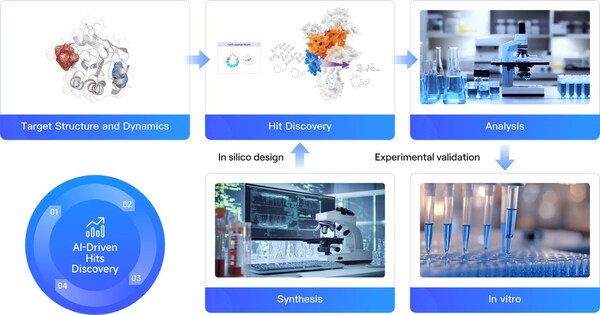
Research on ferroptosis is gaining momentum, but the development of small molecule inhibitors faces numerous challenges
BEIJING, Aug. 1, 2024 /PRNewswire/ -- Glutathione peroxidase 4 (GPX4) is recognized as a critical regulator of ferroptosis, playing a significant role in lipid and amino acid metabolism, as well as influencing cellular aging, oncogenesis, and cell death [1]. Targeting GPX4-mediated ferroptosis presents a promising therapeutic strategy, particularly in the treatment of cancer [2].
Despite its potential, GPX4's flat surface presents significant challenges, as it lacks distinct druggable pockets (Fig. 1A). Current inhibitors of GPX4 with cellular activity typically covalently bind to the selenocysteine residue at position 46, leading to poor selectivity and high toxicity (Fig. 1B).
![Fig. 1 Crystal structure of GPX4 protein in complex with ML162[3] (PDB ID: 6HKQ) and existing covalent inhibitors.](https://mma.prnasia.com/media2/2473521/image_5027215_10170451.jpg?p=medium600 "Fig. 1 Crystal structure of GPX4 protein in complex with ML162[3] (PDB ID: 6HKQ) and existing covalent inhibitors.")
Fig. 1 Crystal structure of GPX4 protein in complex with ML162[3] (PDB ID: 6HKQ) and existing covalent inhibitors.
Given this background, it is crucial to explore potential cryptic pockets or allosteric sites on GPX4 that could influence its biological function.
AI-Powered RiDYMO Platform Identifies Novel Binding Sites and Non-covalent Inhibitors
"Our strategy for developing GPX4 inhibitors centers on identifying cryptic pockets that offer improved druggability," stated Dr. Xiaomin Zhang, Head of Drug Discovery at DP Technology. "The company's AI for Science-based RiDYMO platform employs molecular dynamics simulations to reveal conformational changes within proteins, facilitating the discovery of novel druggable pockets. The non-covalent molecules developed using these pockets demonstrate enzyme inhibition and cellular activity comparable to established covalent controls, while exhibiting improved druggability. This highlights the RiDYMO platform's significance in drug discovery, particularly for challenging targets."
The workflow utilized by DP Technology's research team for this project encompassed the following steps:
1. Protein Conformation Sampling: The GPX4 protein was simulated using Reinforced Dynamics (RiD)[4]. As illustrated in Fig. 2, RiD surpasses traditional Molecular Dynamics (MD) by more effectively exploring the protein's conformational space, revealing additional metastable conformations and cryptic sites (with pink areas indicating amino acids concealed within the protein).

Fig. 2 Enhanced Sampling of GPX4 Protein with RiD
2. Induction of Druggable Pockets: After identifying hidden sites, we conducted further exploration and induction of these pockets using organic solvent probes. As shown in Fig. 3, these organic solvent probes facilitated the formation of deeper small molecule binding pockets on the previously flat surface of the protein.

Fig. 3 Molecular dynamics simulations combined with the organic solvent probe to induce the discovery of a new pocket in GPX4.
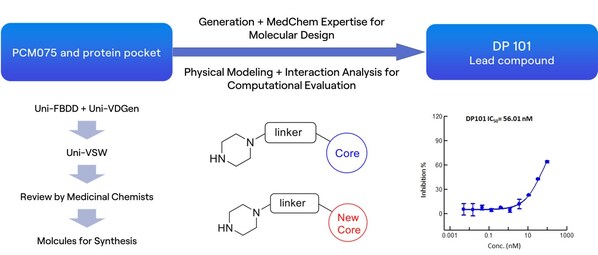
3. Molecule Screening and Validation: High-throughput virtual screening was conducted on these pockets using the Uni-Dock[5] from DP Technology's Hermite drug design platform. The non-covalent molecules DP018 and DP029 exhibited micromolar-level inhibition of GPX4 enzymatic activity (Fig. 4A), comparable to the covalent inhibitor ML162. Surface Plasmon Resonance (SPR) experiments confirmed the direct binding of non-covalent molecules to GPX4 with micromolar affinity (Fig. 4B). Additionally, results from the ROS assay (Fig. 4C) indicate that these non-covalent molecules effectively induce ferroptosis in cells.
, SPR assay (B), and ROS assay (C)")
Fig. 4 Results of ML162 and screened molecules in GPX4 enzymatic assay (A), SPR assay (B), and ROS assay (C)
4. Summary: James et al. highlighted four limitations of structural biology in drug development in their publication in Cell [6]. One of these challenges is that protein wiggling and jiggling are crucial but challenging to model experimentally and computationally, future drug design will unlock methods based on the ensemble of structures. In our research, we conducted a comprehensive analysis of the dynamics of the protein's backbone and side chains using RiD simulation and organic solvent probing. By effectively identifying two druggable pockets and conducting high-throughput virtual screening of compound libraries totaling approximately ten million compounds, we discovered around ten active non-covalent inhibitor molecules. Notably, two of these inhibitors demonstrated high enzymatic activity and strong binding affinity, with their capacity to induce ferroptosis further validated at the cellular level.

Fig. 5 The workflow of identifying novel non-covalent hits against GPX4 using the RiDYMO® Reinforced Dynamics Platform
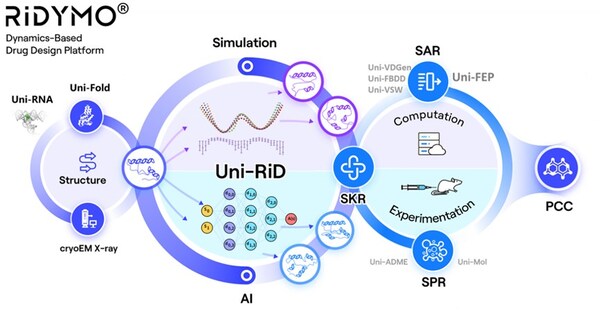
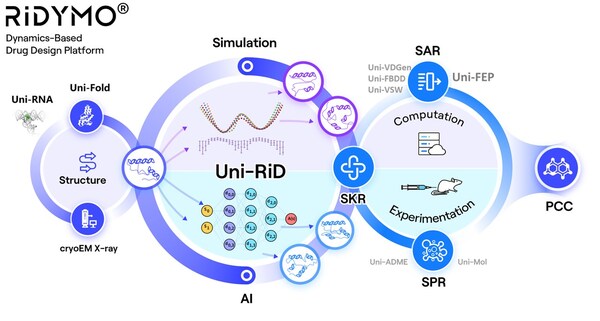
Making Proteins "Move" – The RiDYMO® Reinforced Dynamics Platform
The RiDYMO® platform integrates various AI and physical algorithms, dedicated to the development of drugs for "undruggable" targets and "best-in-class" molecules. As one of its core algorithms, Reinforced Dynamics (RiD)[4] has a significant advantage in the sampling efficiency of molecular dynamics simulation. By fully leveraging the high-dimensional representation capabilities of neural networks, RiD can efficiently capture dynamic conformational changes in complicated biomolecular systems. Previously, the core RiD algorithm of the platform was published in Nature Computational Science. The study demonstrated that RiD could achieve a more comprehensive free energy surface within 1.86 μs, compared to 100 μs required by traditional MD methods, representing nearly a hundredfold increase in efficiency.
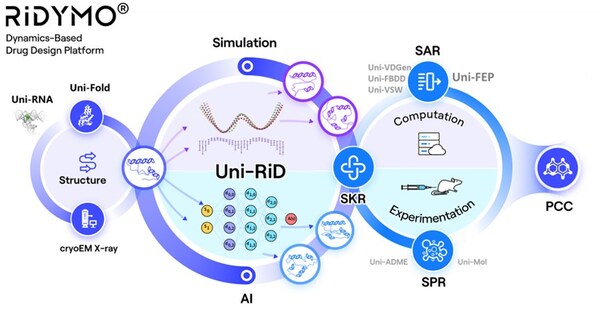
The RiDYMO® platform studies the dynamics of biological systems, revealing cryptic binding sites across challenging systems, including protein-protein interactions (PPIs), intrinsically disordered proteins (IDPs), membrane proteins, and RNA. Its effectiveness has been validated on targets such as the COPI protein, MCL1 protein, c-Myc protein, GPX4 protein, NMDA protein, Nav1.8 protein, and c-Myc RNA.
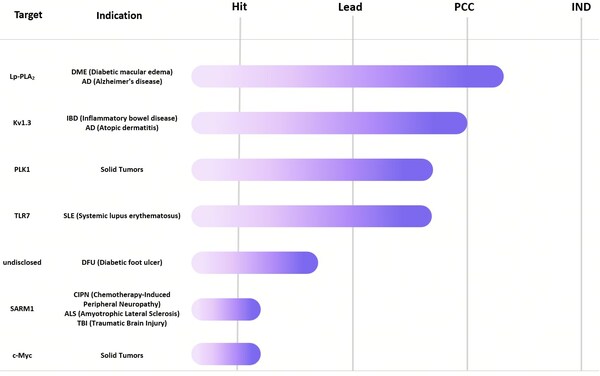
We look forward to collaborating with trusted partners to advance these initiatives through all stages of development, including hit, lead, PCC, and IND. For collaboration inquiries or additional information, please contact bd@dp.tech.
About DP Technology
DP Technology is a global leader in the "AI for Science" research paradigm, where AI learns scientific principles and data, then tackles key challenges in scientific research and industrial R&D.
We've developed the "DP Particle Universe," a suite of advanced pre-trained models that seamlessly connect cutting-edge research with real-world industrial applications. Our software suite includes:
Together, these platforms form a robust foundation for industrial innovation and an open ecosystem for AI in science, fostering advancements in key areas such as drug discovery, energy, material science, and information technology.
Relying on DP Technology's advanced Reinforced Dynamics Platform, RiDYMO®, we have set up a world-leading hit discovery system. The team has established external collaborations and built up a strong internal pipeline, focusing on three areas: CNS, oncology, and autoimmune diseases.
Reference
[1] Stockwell, B. R. (2022). Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell, 185(14), 2401-2421. [2] Wang, F., & Min, J. (2021). DHODH tangoing with GPX4 on the ferroptotic stage. Signal Transduction and Targeted Therapy, 6(1), 1-2. [3] Moosmayer, D., Hilpmann, A., Hoffmann, J., Schnirch, L., Zimmermann, K., Badock, V., ... & Hillig, R. C. (2021). Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallographica Section D: Structural Biology, 77(2), 237-248. [4] Wang, D., Wang, Y., Chang, J., Zhang, L., Wang, H. & E, W. (2022). Efficient sampling of high-dimensional free energy landscapes using adaptive reinforced dynamics. Nature Computational Science, 2(1), 20-29. [5] Yu, Y., Cai, C., Wang, J., Bo, Z., Zhu, Z., & Zheng, H. (2023). Uni-Dock: GPU-accelerated docking enables ultralarge virtual screening. Journal of chemical theory and computation, 19(11), 3336-3345. [6] Fraser, J. S., & Murcko, M. A. (2024). Structure is beauty, but not always truth. Cell, 187(3), 517-520. |